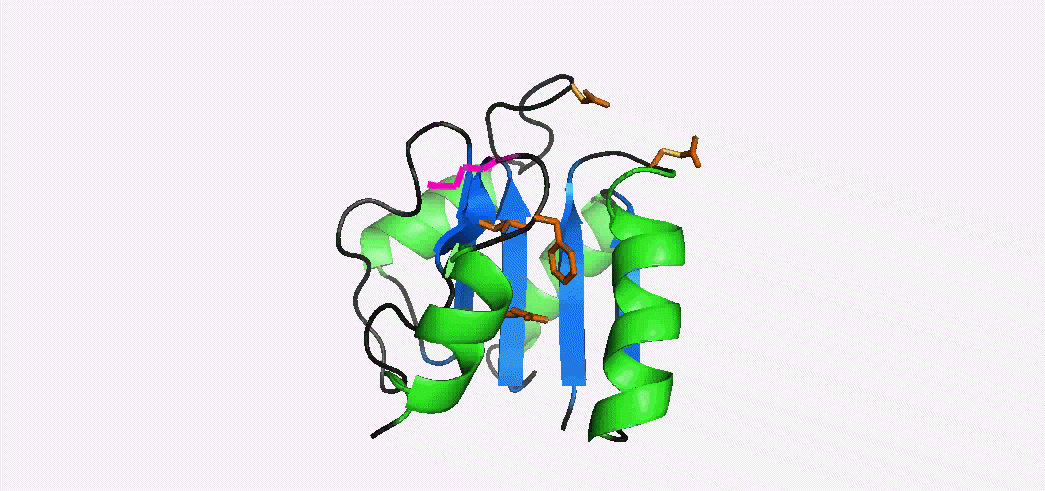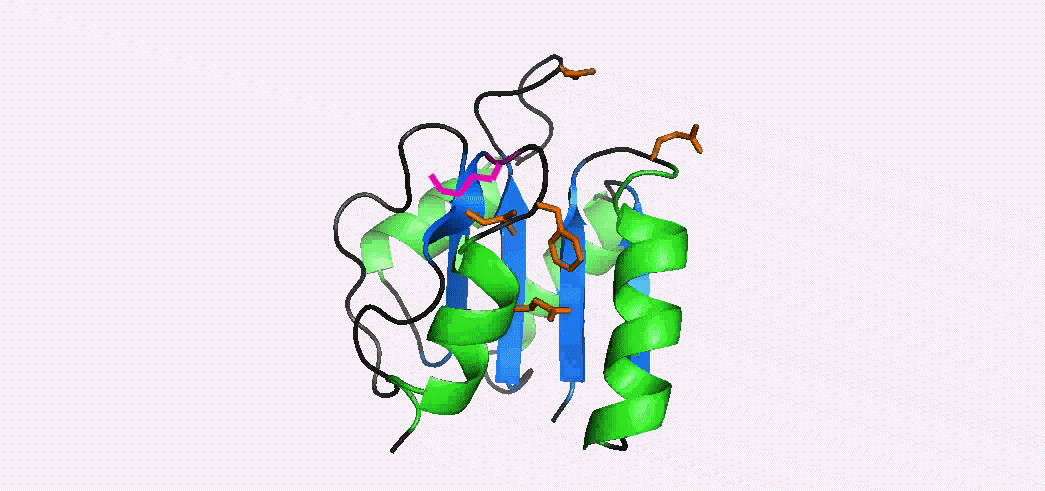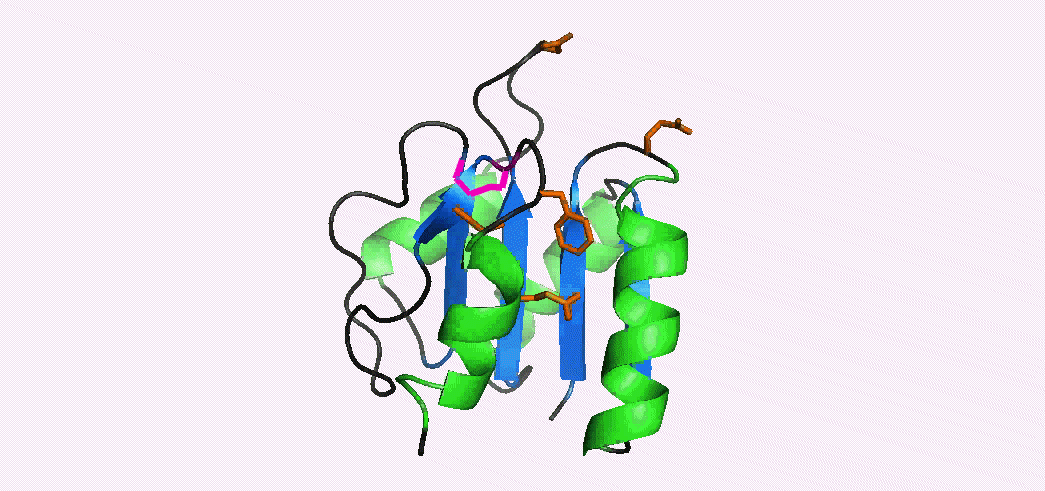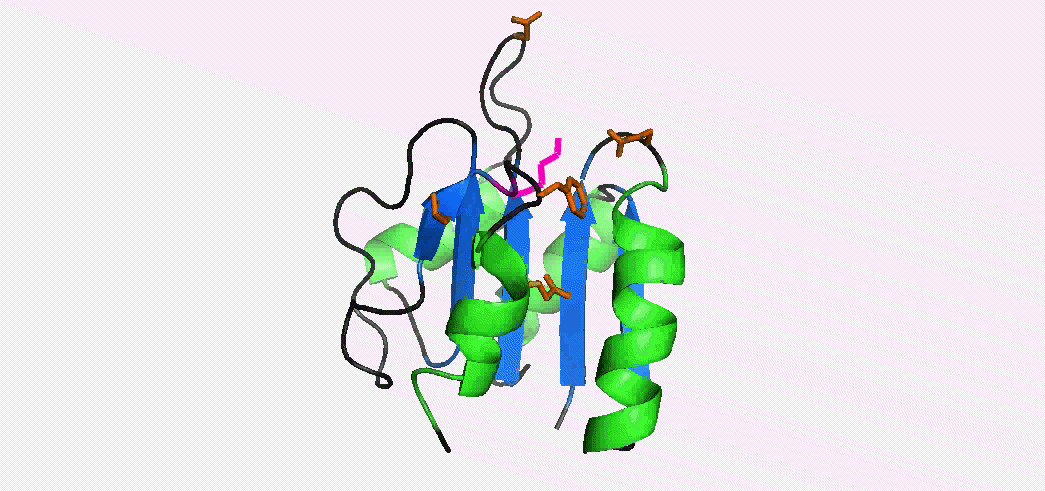“In organello” and “in cellulo” metabolomics
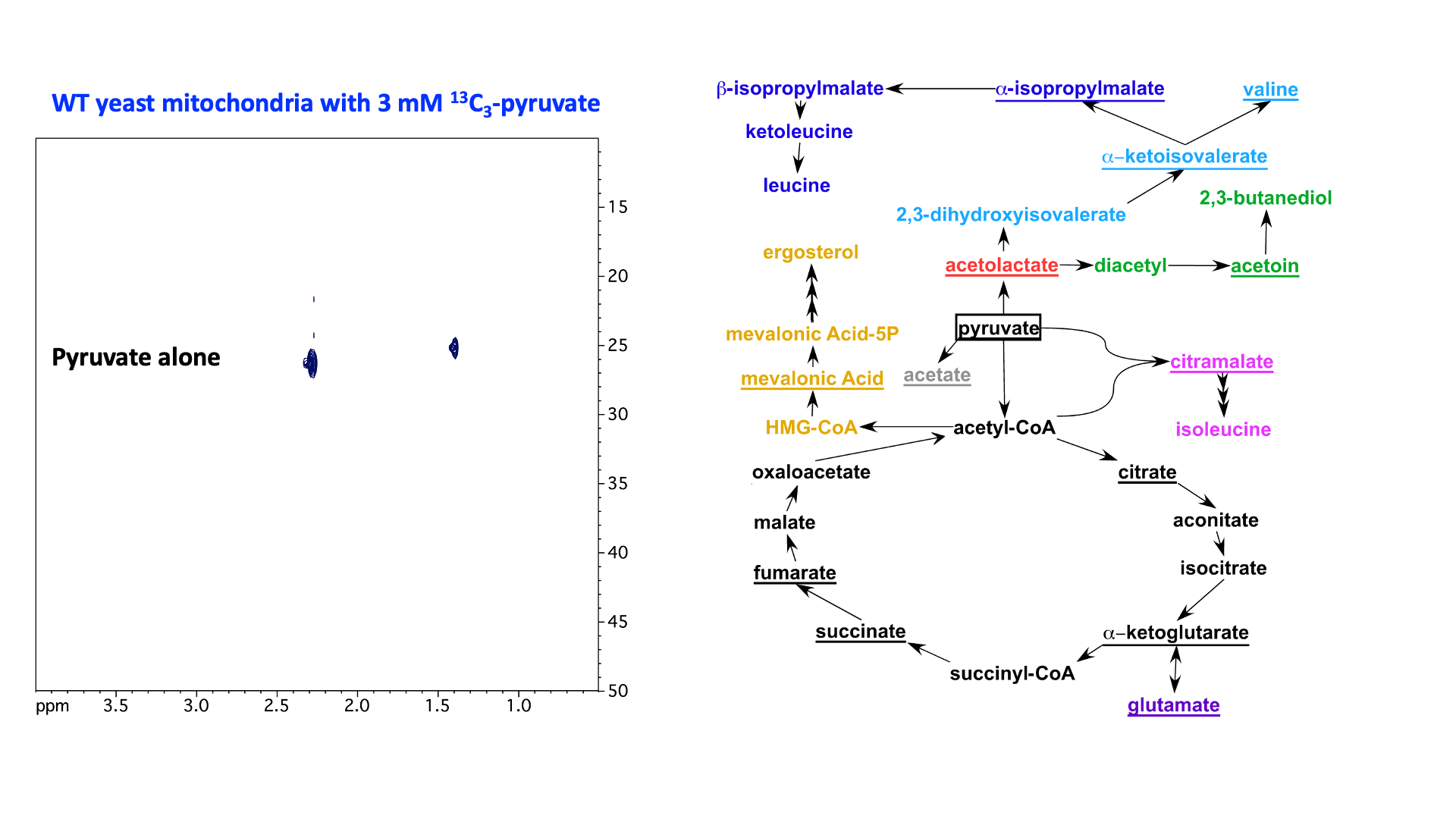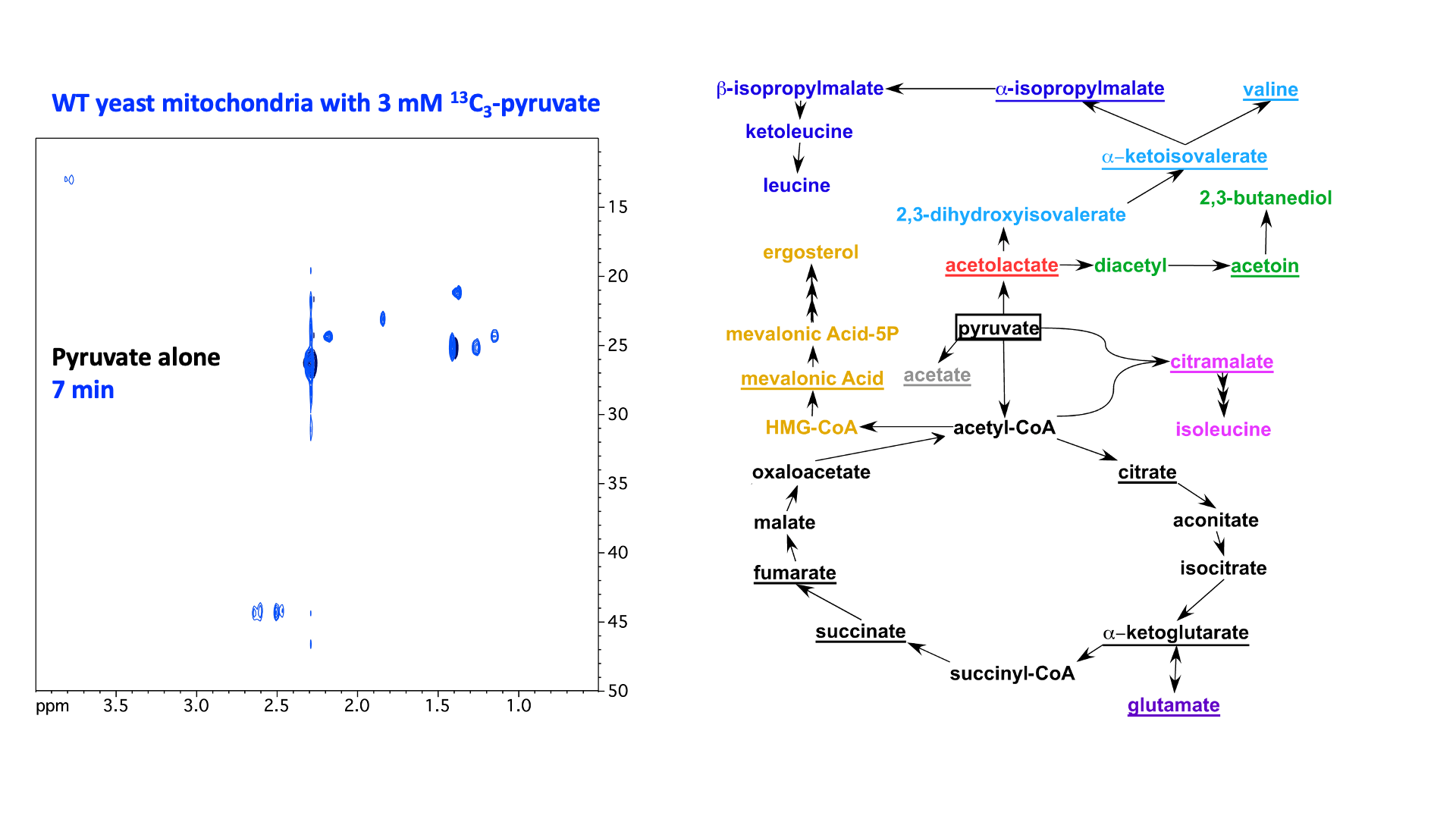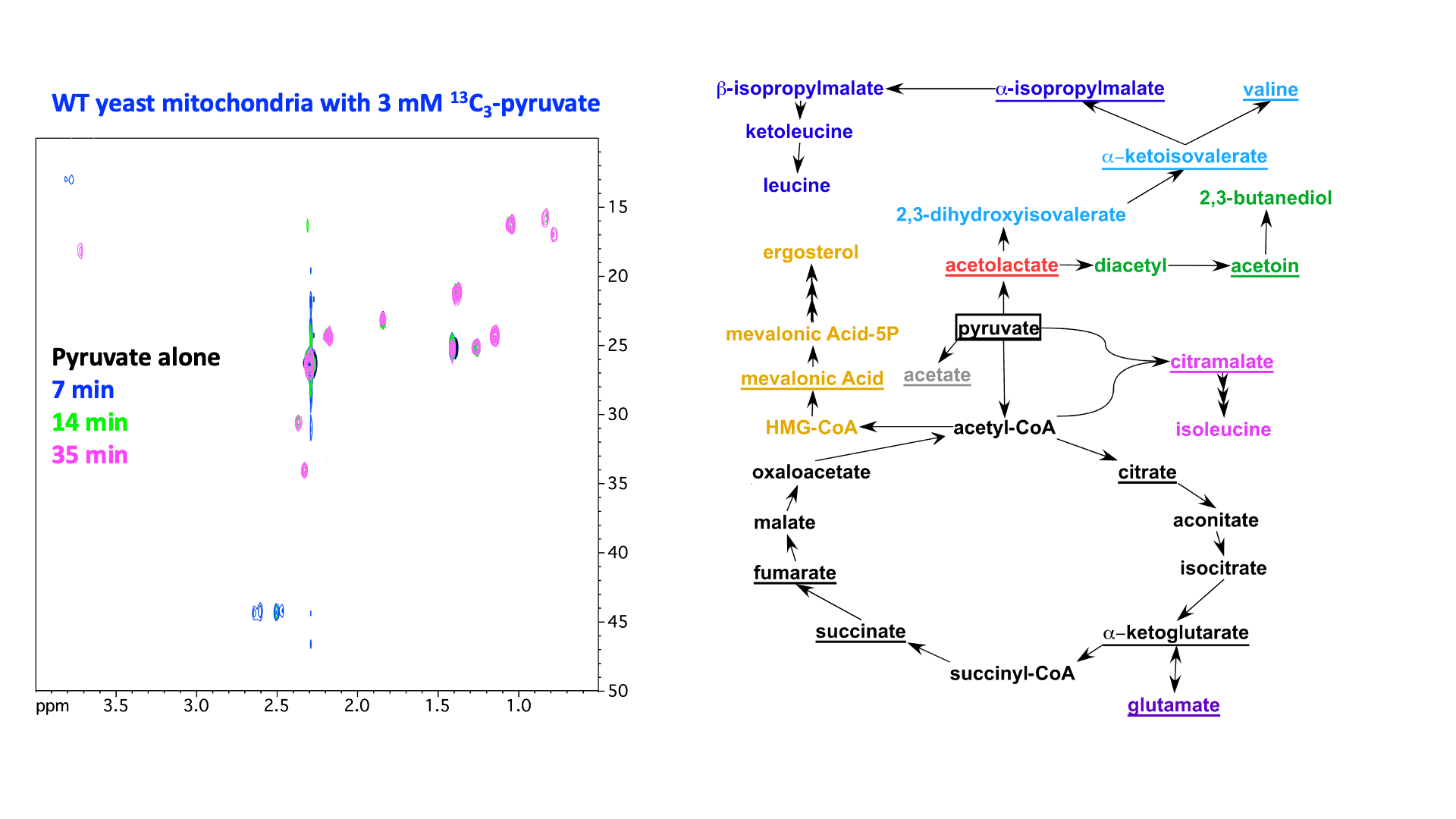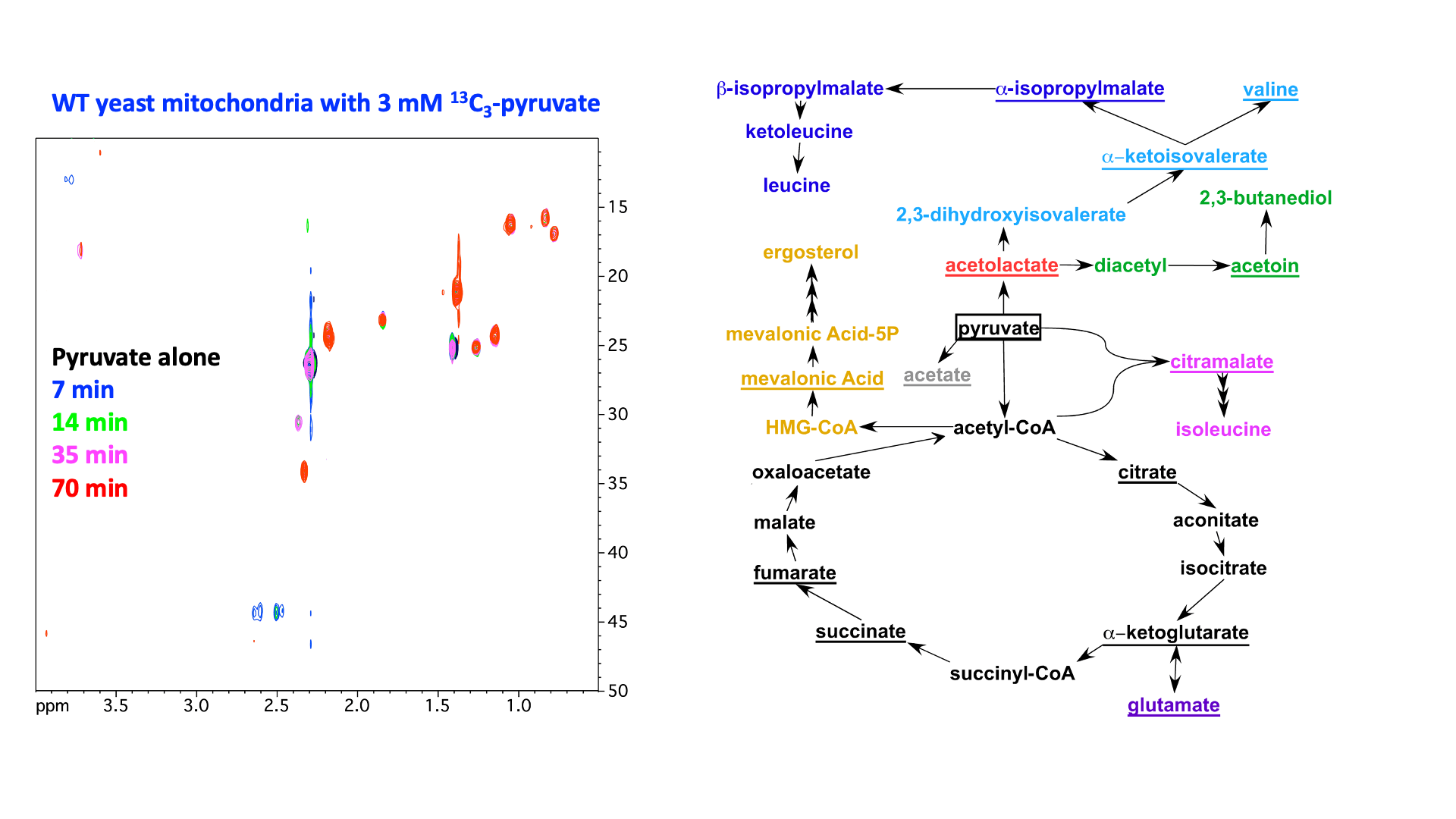
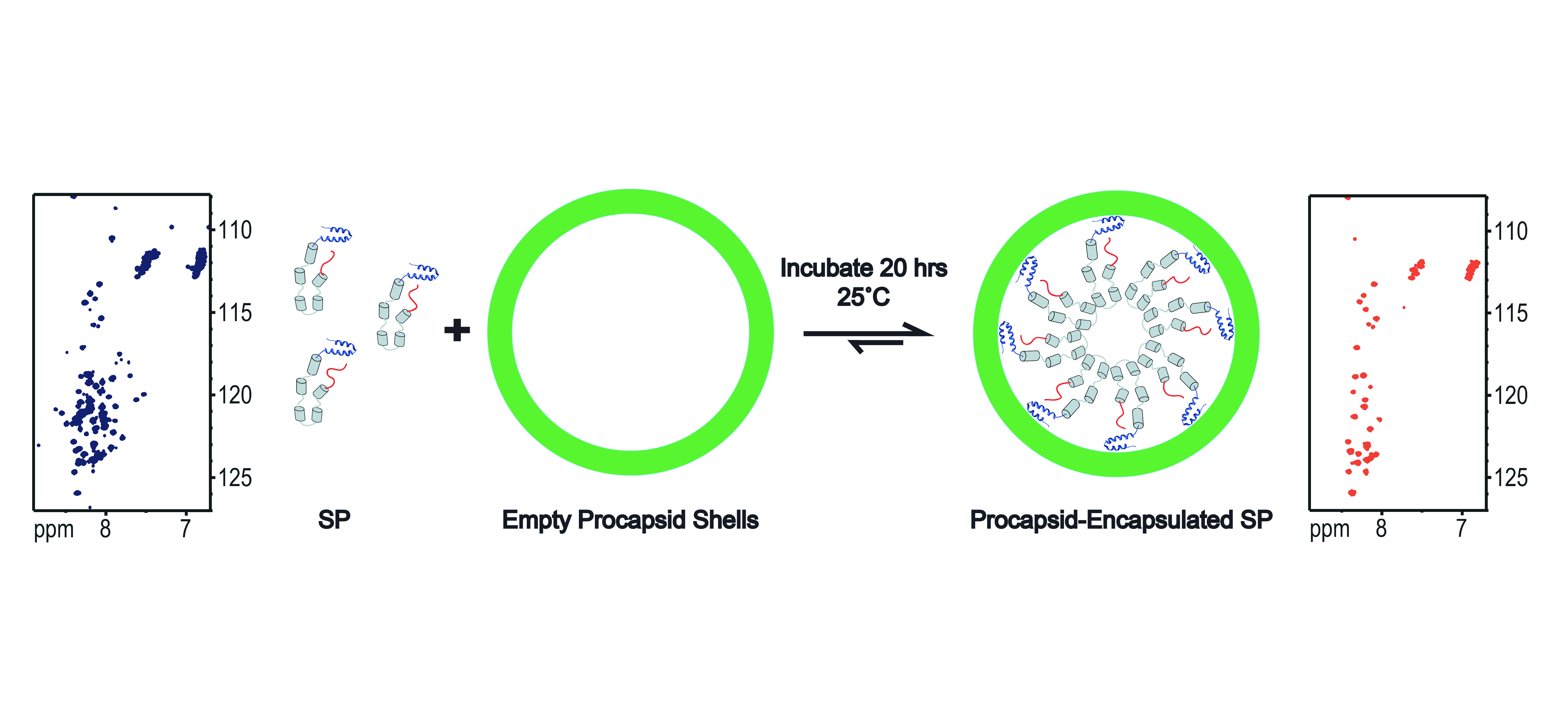
“In Virus” NMR
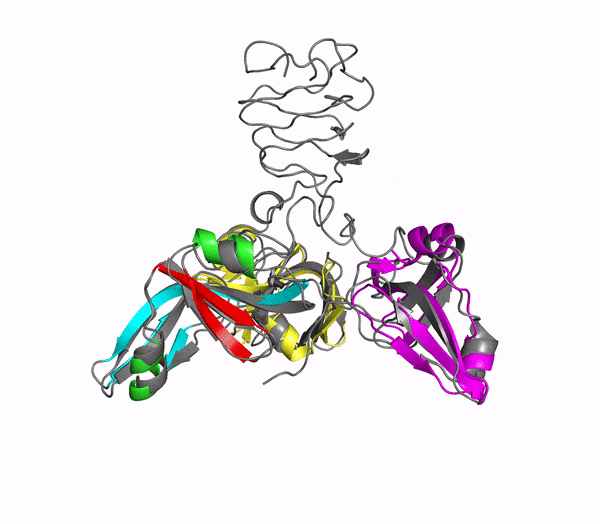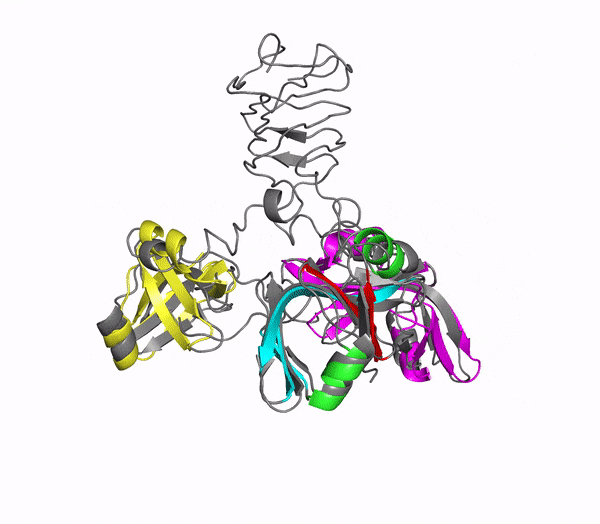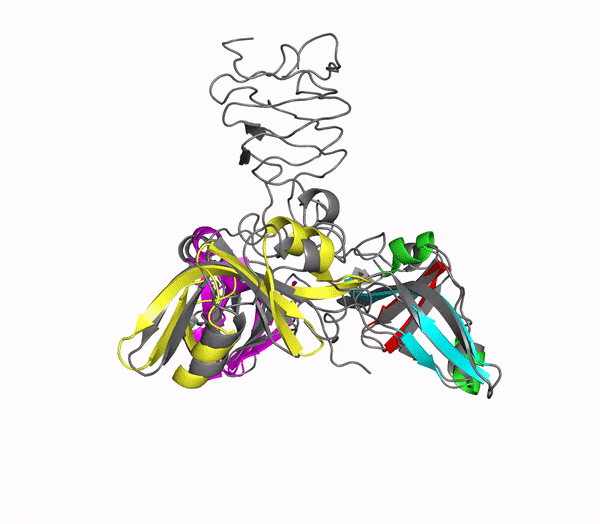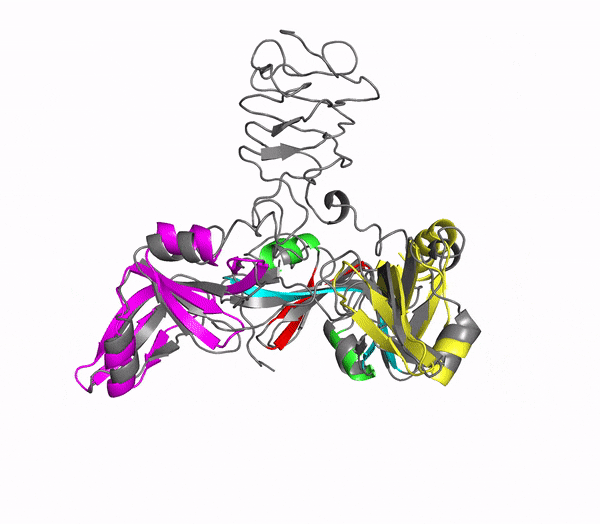
Structure of phage decoration/cementing protein Dec
The C-terminal domain of the bacterial pore-forming-toxin Hemolysin II has a novel fold
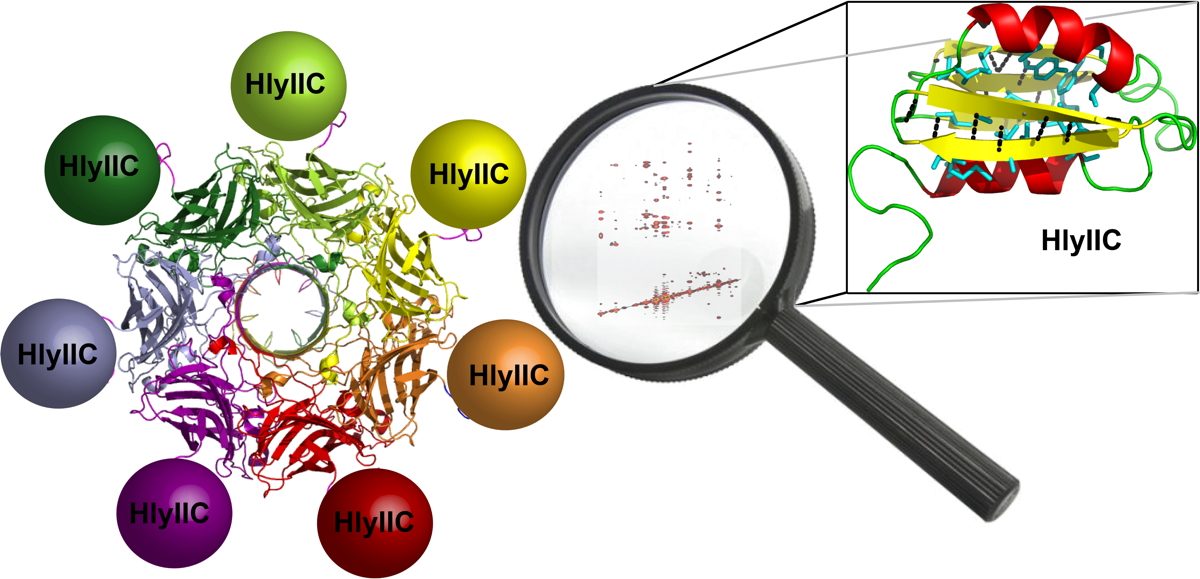
In addition to multiple virulence factors, Bacillus cereus a pathogen that causes food poisoning and life-threatening wound infections, secretes the pore-forming toxin hemolysin II (HlyII). Pore-forming-toxins (PFTs) are membrane-spanning oligomers that typically perforate target cell membranes releasing their nutrients. The HlyII toxin has a unique 94 amino acid C-terminal domain (HlyIIC). In collaboration with Rich Olson's lab at Wesleyan University, we have determined the NMR structure of the HlyIIC domain. HlyIIC exhibits splitting of NMR resonances due to cis/trans isomerization of a single proline near the C-terminus. To overcome heterogeneity, we solved the structure of P405M-HlyIIC, a mutant that exclusively stabilizes the trans state. The NMR structure of HlyIIC reveals a novel fold, consisting of two subdomains αA-β1-β2 and β3-β4-αB-β5, that come together in a barrel-like structure. The barrel core is fastened by three layers of hydrophobic residues. The barrel end opposite the HlyIIC-core has a positively charged surface, that by binding negatively charged moieties on cellular membranes, may play a role in target-cell surface recognition or stabilization of the heptameric pore complex. In the destabilizing P405M mutant, increased flexibility is evident throughout the first subdomain, suggesting that the HlyIIC structure may have arisen through gene fusion. Current work is aimed at characterizing two minor conformational forms of HlyIIC, that will lead to a more comprehensive understanding of the structural properties of this novel domain.
Link to the Olson's group at Weslyan University
Kaplan, A.R, Kaus, K., De, S., Olson, R., & Alexandrescu, A.T. (2017) “NMR structure of the Bacillus cereus hemolysin II C-terminal domain reveals a novel fold”, Scientific Reports 7, 3277.
Kaplan, A.R., Maciejewski, M.W., Olson, R., & Alexandrescu, A.T. (2014) “NMR assignments for the cis and trans forms of the hemolysin II C-terminal domain”. Biomolecular NMR Assignments 8, 419-423.
Structures of Insertion (I)-domains in phage coat proteins
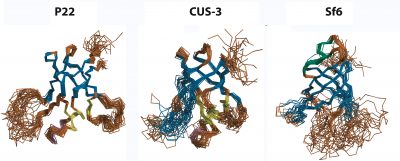 Viruses are self-contained infectious agents that target organisms ranging from animals and plants to bacteria and Archaea. Viruses that specifically target bacteria, called bacteriophages or phages, such as phage P22 which infects Salmonella typhimurium, are ubiquitous in nature and share many of the properties and replication mechanisms of viruses important in human diseases such as herpesvirus. Icosahedral viruses encode coat proteins that form a capsid, to protect their genomes from the environment and host defenses. The coat proteins of a number of phages and viruses are built around a conserved "HK97-fold", named after the Hong Kong 97 virus, the first example of this subfamily to have its coat protein structure determined. The coat proteins of some viruses with the HK97-fold have additional accessory domains that impart specific functions. In collaboration with Carol Teschke's group we have characterized the structure, dynamics and biophysics of the non-conserved I-domain insertion of the phage P22 coat protein. The NMR structure of the I-domain shows that it folds into an anti-parallel 6-stranded β-barrel fold, previously not observed in HK97-fold accessory domains. The D-loop, which is dynamic both in the isolated I-domain and in the intact monomeric coat protein, forms stabilizing salt bridges between adjacent capsomers in procapsids. A newly described S-loop is important for capsid size determination, likely through intra-subunit interactions. The I-domain has a markedly asymmetric distribution of charges, where negatively charged acidic groups are positioned on the surface of the capsid shell and positively charged basic groups dock the module against a negatively charged patch on the core HK97-fold of the coat protein. Ten of eighteen coat protein temperature-sensitive-folding (tsf) substitutions, which restrict phage infectivity at non-permissive temperatures, are located within the I-domain, indicating its importance in coat protein folding and stability. Several of these tsf mutations are found on the positively charged face of the β-barrel that anchors the I-domain to a negatively charged surface of the coat protein HK97-core. The I-domain thus appears to have multifunctional roles in the P22 coat protein that include nucleating the folding of the coat protein, modulating the stability of the coat protein, forming intermolecular interactions important in capsid assembly, as well as possibly interacting with the portal protein. Future work will focus on further defining the folding and stability of the I-domain, better defining the roles of electrostatic interactions, as well as NMR and cryo-EM studies of non-conserved accessory domains from other phages that will shed light on whether the I-domains have conserved structures and functions in phages and viruses based on the HK97-fold. We have also looked into the question of how well-conserved the I-domain is, by determining structures from the three phages P22, CUS-3, and Sf6, that represent the three main clusters in a family of 151 dsDNA phages.
Viruses are self-contained infectious agents that target organisms ranging from animals and plants to bacteria and Archaea. Viruses that specifically target bacteria, called bacteriophages or phages, such as phage P22 which infects Salmonella typhimurium, are ubiquitous in nature and share many of the properties and replication mechanisms of viruses important in human diseases such as herpesvirus. Icosahedral viruses encode coat proteins that form a capsid, to protect their genomes from the environment and host defenses. The coat proteins of a number of phages and viruses are built around a conserved "HK97-fold", named after the Hong Kong 97 virus, the first example of this subfamily to have its coat protein structure determined. The coat proteins of some viruses with the HK97-fold have additional accessory domains that impart specific functions. In collaboration with Carol Teschke's group we have characterized the structure, dynamics and biophysics of the non-conserved I-domain insertion of the phage P22 coat protein. The NMR structure of the I-domain shows that it folds into an anti-parallel 6-stranded β-barrel fold, previously not observed in HK97-fold accessory domains. The D-loop, which is dynamic both in the isolated I-domain and in the intact monomeric coat protein, forms stabilizing salt bridges between adjacent capsomers in procapsids. A newly described S-loop is important for capsid size determination, likely through intra-subunit interactions. The I-domain has a markedly asymmetric distribution of charges, where negatively charged acidic groups are positioned on the surface of the capsid shell and positively charged basic groups dock the module against a negatively charged patch on the core HK97-fold of the coat protein. Ten of eighteen coat protein temperature-sensitive-folding (tsf) substitutions, which restrict phage infectivity at non-permissive temperatures, are located within the I-domain, indicating its importance in coat protein folding and stability. Several of these tsf mutations are found on the positively charged face of the β-barrel that anchors the I-domain to a negatively charged surface of the coat protein HK97-core. The I-domain thus appears to have multifunctional roles in the P22 coat protein that include nucleating the folding of the coat protein, modulating the stability of the coat protein, forming intermolecular interactions important in capsid assembly, as well as possibly interacting with the portal protein. Future work will focus on further defining the folding and stability of the I-domain, better defining the roles of electrostatic interactions, as well as NMR and cryo-EM studies of non-conserved accessory domains from other phages that will shed light on whether the I-domains have conserved structures and functions in phages and viruses based on the HK97-fold. We have also looked into the question of how well-conserved the I-domain is, by determining structures from the three phages P22, CUS-3, and Sf6, that represent the three main clusters in a family of 151 dsDNA phages.
Tripler, T.N., Kaplan, A.R., Alexandrescu, A.T. & Teschke, C.M. (2019) “Conservation and divergence of the I-domain inserted into the ubiquitous HK97 coat protein fold in the P22-like bacteriophages.” Journal of Virology 93, iii: e0007-19.
Tripler, T.N., Teschke, C.M. & Alexandrescu, A.T. (2017) “NMR assignments for the insertion domain of bacteriophage Sf6 coat protein.” Biomolecular NMR Assignments 11, 35-38.
Harprecht, C., Okifo, O., Robbins, K.J., Motwani, T., Alexandrescu, A.T. & Teschke, C.M. & (2016) “Contextual role of a salt-bridge in the phage P22 coat protein I-domain.”, J. Biol. Chem. 291, 11359-11372.
Newcomer, R.L., Fraser, L.C.R., Teschke, C.M. & Alexandrescu, A.T. (2015) “Mechanism of protein denaturation: partial unfolding of the P22 coat protein I-domain by urea binding.”, Biophysical J. 109, 2666-2677.
Tripler, T.N., Maciejewski, M.W., Teschke, C.M. & Alexandrescu, A.T. (2015) “NMR assignments for the insertion domain of bacteriophage CUS-3 coat protein.” Biomolecular NMR Assignments 9, 333-336.
Rizzo, A.A., Suhanovsky, M.M., Baker, M.L., Fraser, L.C.R., Jones, L.M., Rempel, D.L., Gross, M.L., Chiu, W., Alexandrescu, A.T. & Teschke, C.M. (2014) “Multiple functional roles of the accessory I-domain of bacteriophage P22 coat protein revealed by NMR structure and cryoEM modeling”. Structure 22, 830-841.
Rizzo, A.A., Fraser, L.C.R., Sheftic, S.R., Suhanovsky, M.M., Teschke, C.M. & Alexandrescu, A.T. (2013) “NMR assignments for the telokin-like domain of bacteriophage P22 coat protein.” Biomolecular NMR Assignments 7, 257-260.
Inhibition of amyloid fibril growth by electrostatic repulsion between like-charges

Consideration of amyloid structures suggests that like-charges, replicated along the one-dimensional lattice that constitutes the fibril axis, should be one of the main forces opposing fibril assembly. To probe the role of electrostatic repulsion in fibrillogenesis we have studied the pH-dependence of a number of amyloidogenic proteins including A-beta (involved in Alzheimer's disease), alpha-synuclein (involved in Parkinson's disease), and amylin (involved in type 2 diabetes). Amylin is a particularly suitable system, since it only has two ionizable sites: the alpha-amino group at the N-terminus and His18 in the beta-sheet core of the amyloid structure. Our approach to determining the pKa values (ionization constants) for these sites has been to look at the pH-dependence of fibrillization kinetics in amylin variants that have only one of the two groups. The α-amino group at the unstructured N-terminus of amylin has a pKa near 8.0, similar to the value in random coil models, and makes relatively small contributions to fibrilogenesis. By contrast, His18, which participates in the intermolecular β-sheet structure of the fibrils, has a pKa lowered to 5.0 in the fibrils compared to the random coil value of 6.5. The lowered pKa of His18 is due to the hydrophobic environment of the residue, and electrostatic repulsion between positively charged His18 residues on neighboring amylin molecules in the fibril. His18 acts as an electrostatic switch, inhibiting fibrillization in its charged state at acidic pH and favoring fibrillization in its uncharged state at neutral pH. The presence of a charged side-chain at position 18 also affects fibril morphology and lowers amylin cytotoxicity towards a MIN6 mouse model of pancreatic β-cells. We are currently exploiting the principle that electrostatic repulsion interferes with fibril formation, to design peptide variants that incorporate strings of like-charged residues in the amylin amino acid sequence. We hope some of these peptides will inhibit amylin fibrillogenesis and cytotoxicity.
Patil S.M., & Alexandrescu, A.T. (2015) “Charge-based inhibitors of amylin fibrillization and totoxicity.” J. Diabetes Res, Article ID 946037.
Jha, S., Snell, J.M., Sheftic, S.R., Patil S.M., Daniels, S.B., Kolling, F.W., & Alexandrescu, A.T. (2014) “pH dependence of amylin fibrillization”. Biochemistry 53, 300-310.
Croke, R.L., Patil, S.M., Quevreaux, J., Kendall, D.A. & Alexandrescu, A.T. (2011) “NMR determination of pKa values in α-synuclein”. Protein Science 20, 256-269.
Sheftic, S.R., Croke, R.L., LaRochelle, J.R., & Alexandrescu, A.T. (2009) “Electrostatic contributions to the stabilities of native proteins and amyloid complexes.” Methods in Enzymology 466, 233-258.
NMR quenched hydrogen exchange of amyloid fibrils
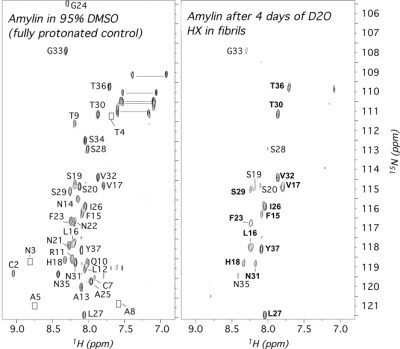 Amyloidogenic proteins and peptides are associated with over 30 human diseases. In amyloid fibrils, the polypeptide chain folds into a highly stable intermolecular cross β-sheet structure that is highly resistant to most denaturants and proteases. These aggregates are extremly challenging to study by most structural methods, particularly solution NMR. Quenched hydrogen exchange (qHX) experiments can circumvent some of the limitations solution NMR faces with amyloid fibrils, namely the large sizes of the aggregates and their insolubility, by transferring information on amide proton occupancy in the fibril to the denatured state. In the qHX experiment, hydrogen exchange (HX) is initiated by suspending amyloid fibrils in D2O. After varying periods of time, HX is quenched by flash-freezing. The partially exchanged fibril samples are lyophilized and dissolved in a strongly denaturing solvent such as 95% dimethyl sulfoxide (DMSO). The DMSO solvent serves two purposes. First, DMSO is sufficiently chaotropic to unfold most types of amyloid fibrils to monomers. Second, because DMSO is an aprotic solvent, HX from the denatured state occurs on timescales of hours, allowing the detection of amide protons trapped in the fibril. The qHX technique was first described for model amyloid fibrils formed by the Escherichia coli protein CspA. Since the method was first published in 2001, it has been used to study a number of amyloid fibrils relevant to human disease. To date the method has been used mainly to investigate the secondary structures of proteins in amyloid fibrils, since hydrogen-bonded secondary structure protects amide protons from solvent exchange. The method, however, also has the potential to investigate whether protection in fibrils accrues through intermediates or arises in an all-or-none fashion, to look at how fibril structure changes with solution conditions, and to determine binding sites for ligands that could be developed into drugs to target fibril growth.
Amyloidogenic proteins and peptides are associated with over 30 human diseases. In amyloid fibrils, the polypeptide chain folds into a highly stable intermolecular cross β-sheet structure that is highly resistant to most denaturants and proteases. These aggregates are extremly challenging to study by most structural methods, particularly solution NMR. Quenched hydrogen exchange (qHX) experiments can circumvent some of the limitations solution NMR faces with amyloid fibrils, namely the large sizes of the aggregates and their insolubility, by transferring information on amide proton occupancy in the fibril to the denatured state. In the qHX experiment, hydrogen exchange (HX) is initiated by suspending amyloid fibrils in D2O. After varying periods of time, HX is quenched by flash-freezing. The partially exchanged fibril samples are lyophilized and dissolved in a strongly denaturing solvent such as 95% dimethyl sulfoxide (DMSO). The DMSO solvent serves two purposes. First, DMSO is sufficiently chaotropic to unfold most types of amyloid fibrils to monomers. Second, because DMSO is an aprotic solvent, HX from the denatured state occurs on timescales of hours, allowing the detection of amide protons trapped in the fibril. The qHX technique was first described for model amyloid fibrils formed by the Escherichia coli protein CspA. Since the method was first published in 2001, it has been used to study a number of amyloid fibrils relevant to human disease. To date the method has been used mainly to investigate the secondary structures of proteins in amyloid fibrils, since hydrogen-bonded secondary structure protects amide protons from solvent exchange. The method, however, also has the potential to investigate whether protection in fibrils accrues through intermediates or arises in an all-or-none fashion, to look at how fibril structure changes with solution conditions, and to determine binding sites for ligands that could be developed into drugs to target fibril growth.
Alexandrescu, A.T. (2016) “Quenched Hydrogen Exchange NMR of Amyloid Fibrils.” Methods in Mol. Biol. 1345, 211-222.
Alexandrescu, A.T. (2013) “Amide proton protection in amylin fibrils probed by quenched hydrogen exchange NMR”, PLoS ONE 8, e56467.
Alexandrescu, A.T. (2001) “An NMR based quenched hydrogen exchange investigation of model amyloid fibrils formed by the protein CspA”, Pac. Symp. Biocomput. 6, 67-78.
Inhibition of SEVI amyloid formation by metals found in seminal fluid
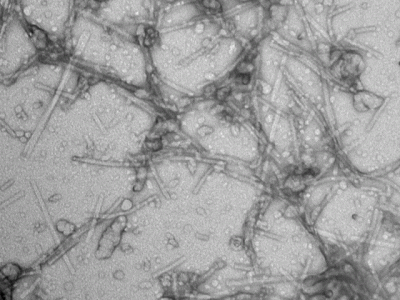
Amyloidogenic proteins assemble into fibrils with a cross β-sheet structure that is involved in a number of human diseases. SEVI is a naturally occurring peptide fragment of human prostatic acid phosphatase, that assembles into amyloid fibrils with a cationic surface. These fibrils enhance HIV fusion of virions with target cells by 6 orders of magnitude by reducing electrostatic repulsion between the negatively charged virus membranes and target cell membranes. As such SEVI amyloids may have a role in HIV transmission. We found that the assembly of SEVI into amyloid fibrils is inhibited by the metals Cu(II) and Zn(II) at concentrations comparable to those found in seminal fluid. The ligands form oligomeric complexes with the SEVI peptide by binding to the two histidines positions 3 and 23 in the amino acid sequence. The oligomeric SEVI complexes with metals are less competent to participate in the nucleation of SEVI fibrils – this is manifested primarily as in increase in the lag time for fibril formation in the presence of metals. Our work suggests that compounds that bind and stabilize non-native structures incompatible with fibril nucleation and/or assembly, may provide an avenue for the development of inhibitors of amyloidogenic polypeptides that are intrinsically unfolded under physiological conditions.
Sheftic, S.R., Snell, J. M., Jha, S. & Alexandrescu, A.T. (2012) “Inhibition of Semen-derived Enhancer of Virus Infection (SEVI) fibrillogenesis by physiological zinc and copper concentrations”, European Biophysics Journal 41, 695-704.
Imaging amyloid fibril growth by Total Internal Reflection Fluorescence (TIRF) microscopy
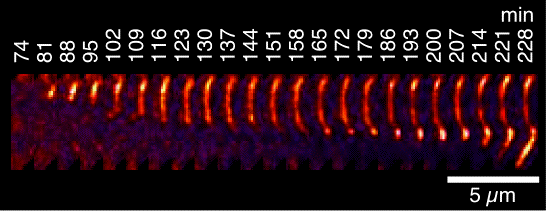 We have been using TIRF microscopy (a variant of fluorescence microscopy) to image the growth of individual amylin fibrils along the plane of a slide. A movie of a fibrillization reaction is shown on our main page - its rather long at about 5 minutes compressed from data collected over 16 h. The random speckle streaks that appear in the movie are from conversion to a GIF file (sorry). TIRF microscopy studies pioneered by the Goto lab have provided information on the mechanism and polarity of fibril growth. In the case of amylin, we found three types of aggregate growth. In the earliest stages globular seeds are formed. The dimensions of the globular seeds as well as their staining with the amyloid-specific dye thioflavin T indicate that they are plaques of short fibrils. The next species observed are fibrils that invariably grow from large globular seeds or smaller punctate granules. Fibril elongation appears to be unidirectional, although in some cases multiple fibrils radiate from a single seed or granule. An example of unidirectional fibril growth is shown in the figure at the left (if you look carefully the bright granule at the top appears to be moving up in the image but that's from movement of the slide by ~0.5 µM over 4 hr, as the fibril is growing down). After fibrils are formed, some show an increase in fluorescence intensity that we attribute to the growth of new fibrils alongside those previously formed. All three aggregation processes are suggestive of secondary (heterogeneous) nucleation mechanisms in which nucleation occurs on pre-formed fibrils. Consistently, electron micrographs show changes in fibril morphology well after fibrils are first formed, and the growth processes observed by fluorescence microscopy occur after the corresponding solution reactions have reached an initial apparent plateau. The results highlight the importance of secondary nucleation in the fibrillization of amylin, as this could provide a pathway to continue fibril growth once an initial population of fibrils is established. We are currently using TIRF microscopy to study interactions of fibrils with co-factors found in amyloid deposits and to investigate mechansims of cell toxicity.
We have been using TIRF microscopy (a variant of fluorescence microscopy) to image the growth of individual amylin fibrils along the plane of a slide. A movie of a fibrillization reaction is shown on our main page - its rather long at about 5 minutes compressed from data collected over 16 h. The random speckle streaks that appear in the movie are from conversion to a GIF file (sorry). TIRF microscopy studies pioneered by the Goto lab have provided information on the mechanism and polarity of fibril growth. In the case of amylin, we found three types of aggregate growth. In the earliest stages globular seeds are formed. The dimensions of the globular seeds as well as their staining with the amyloid-specific dye thioflavin T indicate that they are plaques of short fibrils. The next species observed are fibrils that invariably grow from large globular seeds or smaller punctate granules. Fibril elongation appears to be unidirectional, although in some cases multiple fibrils radiate from a single seed or granule. An example of unidirectional fibril growth is shown in the figure at the left (if you look carefully the bright granule at the top appears to be moving up in the image but that's from movement of the slide by ~0.5 µM over 4 hr, as the fibril is growing down). After fibrils are formed, some show an increase in fluorescence intensity that we attribute to the growth of new fibrils alongside those previously formed. All three aggregation processes are suggestive of secondary (heterogeneous) nucleation mechanisms in which nucleation occurs on pre-formed fibrils. Consistently, electron micrographs show changes in fibril morphology well after fibrils are first formed, and the growth processes observed by fluorescence microscopy occur after the corresponding solution reactions have reached an initial apparent plateau. The results highlight the importance of secondary nucleation in the fibrillization of amylin, as this could provide a pathway to continue fibril growth once an initial population of fibrils is established. We are currently using TIRF microscopy to study interactions of fibrils with co-factors found in amyloid deposits and to investigate mechansims of cell toxicity.
Patil, S.M., Mehta, A., Jha, S. & Alexandrescu, A. (2011) “Heterogeneous amylin growth mechanisms imaged by Total Internal Reflection Fluorescence Microscopy”. Biochemistry 50, 2808-2819.
Relative stabilities of conserved and non-conserved secondary structure in OB-fold proteins
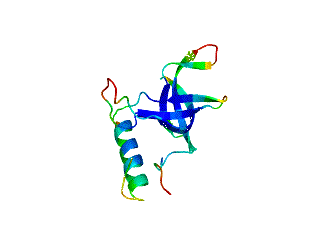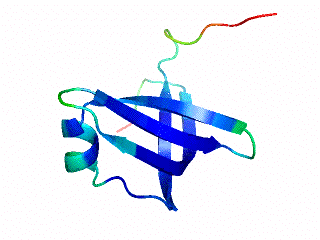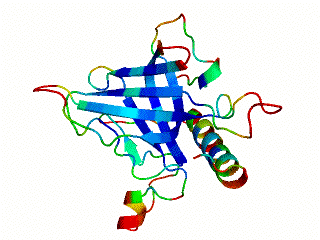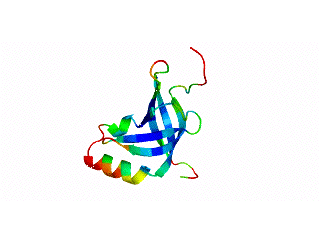
Although many proteins fold in a cooperative two-state transition, other proteins are less cooperatively organized, making it possible to characterize partially folded intermediate states. These partially folded states may offer the best chance to understand protein-folding mechanisms. What accounts for the ability of some proteins to fold to incomplete structures under certain conditions? Our hypothesis is that these partially folded states reflect how the protein structures developed during evolution.
The OB-fold is a diverse structure superfamily based on a five-stranded beta-barrel motif that is often supplemented with additional non-conserved secondary structures. Previous deletion mutagenesis and NMR hydrogen exchange studies of three OB-fold proteins (SN, LysN, CspA) showed that structural stabilities were larger for sites within the conserved beta-barrels than sites in the non-conserved segments of these proteins. To find the physical basis of this behavior we examined a database of 80 representative domain structures currently classified as OB-folds in the SCOP taxonomy. Residue-specific values were obtained for the number of C-alpha to C-alpha distance contacts, sequence hydrophobicities, crystallographic B-factors, and theoretical B-factors calculated from a Gaussian Network Model. All four parameters point to a larger average flexibility for the non-conserved structures compared to the conserved beta-barrels. The theoretical B-factors and contact densities show the highest sensitivity. The figure in the adjacent panel shows the subset of 40 OB-fold domains for which X-ray structures are availale color-coded according to the theoretical B-factors. The color ramp runs from blue (small B-factor, rigid) to red (large B-factor, flexible). For most of the OB-fold structures residues in the beta-barrel are blue, while residues in the non-conserved accessory structures tend towards the red regions of the spectrum.
These results suggest a model of protein structure evolution in which novel structural features develop at the periphery of conserved motifs. Core residues are more resistant to structural changes during evolution since their substitution would disrupt a larger number of interactions. Similar factors probably account for the differences in stability to unfolding between conserved and non-conserved structures.
Guardino, K.M., Sheftic, S.R., Slattery, R.E., & Alexandrescu, A.T. (2009) “Hallmarks of conserved and non-conserved structures in the OB-fold superfamily”. Int. J. Mol. Sci. 10, 2412-2430. (open access, free paper - click here)
Newcomer, R.L., Fraser, L.C.R., Teschke, C.M. & Alexandrescu, A.T. (2015) “Mechanism of protein denaturation: partial unfolding of the P22 coat protein I-domain by urea binding.”, Biophysical J. 109, 2666-2677.
Structure of micelle-bound amylin
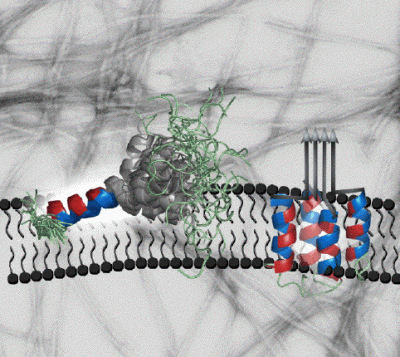 Amylin is a hormone made by the pancreas that regulates food metabolism. Amongst its actions is to induce a feeling of fullness after meals, by binding to receptors in the neurons of the brain's hypothalamus. In addition to it's normal role, amylin is important in the development of type 2 (adult-onset) diabetes - a disease that affects over 100 million people worldwide and costs more than $130 billion a year to treat in the US alone. In the majority (95%) of patients with type 2 diabetes, amylin aggregates into fibrillar amyloid deposits in the pancreas, The deposits form in the extracellular spaces between the beta-cells that make both amylin and insulin. The fibrils, or probably smaller soluble aggregates destroy the beta-cells as type 2 diabetes progresses. The toxic effects of amylin are thought to be exerted through membrane-bound aggregates that form holes in the beta-cell membranes leading to cell death. In order to better understand these processes we used nuclear magnetic resonance (NMR) to determine the structure of amylin in a membrane-like environment. In the presence of SDS micelles (a membrane mimic amenable to NMR studies) amylin folds into an alpha-helix structure. The first 17 residues are partially inserted in the micelle, away from solvent. The segment between amino acids 20-29 that has the strongest inclination to form aggregates is more mobile and is placed at the interface between the micelle and solvent (NMR structures on the left hand side). Our structural model suggests that the positioning and flexibility of the amyloidogenic segment 20-29 promotes the aggregation of amylin on the surfaces of membranes. Aggregation of the 20-29 segment could lead to a repositioning of the first 17 amino acids, from the surface to the interior of the membrane leading to an annular pore structure that is toxic to cells (hypothetical model on the right hand side). Our studies of amylin are still at an early stage. Future goals include to look at the hormone bound to more realistic models of cellular membranes and to structurally characterize the amylin aggregates responsible for cell toxicity
Amylin is a hormone made by the pancreas that regulates food metabolism. Amongst its actions is to induce a feeling of fullness after meals, by binding to receptors in the neurons of the brain's hypothalamus. In addition to it's normal role, amylin is important in the development of type 2 (adult-onset) diabetes - a disease that affects over 100 million people worldwide and costs more than $130 billion a year to treat in the US alone. In the majority (95%) of patients with type 2 diabetes, amylin aggregates into fibrillar amyloid deposits in the pancreas, The deposits form in the extracellular spaces between the beta-cells that make both amylin and insulin. The fibrils, or probably smaller soluble aggregates destroy the beta-cells as type 2 diabetes progresses. The toxic effects of amylin are thought to be exerted through membrane-bound aggregates that form holes in the beta-cell membranes leading to cell death. In order to better understand these processes we used nuclear magnetic resonance (NMR) to determine the structure of amylin in a membrane-like environment. In the presence of SDS micelles (a membrane mimic amenable to NMR studies) amylin folds into an alpha-helix structure. The first 17 residues are partially inserted in the micelle, away from solvent. The segment between amino acids 20-29 that has the strongest inclination to form aggregates is more mobile and is placed at the interface between the micelle and solvent (NMR structures on the left hand side). Our structural model suggests that the positioning and flexibility of the amyloidogenic segment 20-29 promotes the aggregation of amylin on the surfaces of membranes. Aggregation of the 20-29 segment could lead to a repositioning of the first 17 amino acids, from the surface to the interior of the membrane leading to an annular pore structure that is toxic to cells (hypothetical model on the right hand side). Our studies of amylin are still at an early stage. Future goals include to look at the hormone bound to more realistic models of cellular membranes and to structurally characterize the amylin aggregates responsible for cell toxicity
Patil, S.M., Xu, S., Sheftic, S. & Alexandrescu, A. (2009) “Dynamic alpha-helix structure of micelle-bound human amylin”. J. Biol. Chem. 284, 11982-11991.
Charges as modulators of protein structure stability
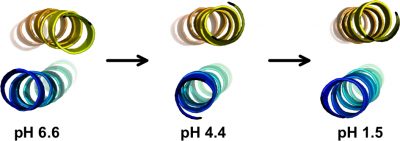
Electrostatic interactions play important roles in protein folding, molecular recognition, and catalytic activity. A better understanding of these forces is important for developing improved potential energy functions, for use in applications such as protein structure prediction and molecular design We obtained a complete set of ionization constants for the GCN4p leucine zipper coiled coil from NMR pH tritrations. This is the first complete set of pKa values of a protein in both its native and denatured states, and serves as a benchmark for testing structure-based electrostatic calculations. In collaboration with the Garcia-Moreno lab at Johns Hopkins, continuum electrostatic calculations were done starting from the GCN4p X-ray structure, to test pKa values and their contributions to protein stability against data from NMR and equilibrium unfolding experiments. The structure-based calculations (dotted red curve) matched the experimentally determined stability profile (blue points), except at the extremes of pH (arrows) where the contributions of charge interactions to stability are greatly overestimated. Part of the reason for the discrepancy may be that the structure or dynamics of the protein change at the extremes of pH and that current theoretical modeling of these changes are inadequate. We are currently using NMR to characterize the structural changes that occur in proteins with changing pH.
A second more recent interest is to look at the roles charges play in the conformational transitions of amyloidogenic proteins. We are investigating the electrostatic properties of amyloidogenic proteins in their monomeric, fibrillar, and micelle-bound states.
Kaplan, A.R., Brady, M.R., Maciejewski, M.W., Kammerer, R.A. & Alexandrescu, A.T. (2017) “NMR Structures of GCN4p are Largely Conserved when Ion Pairs are Disrupted at Acidic pH but Show a Relaxation of the Coiled Coil Superhelix", Biochemistry 56, 1604-1619.
Harprecht, C., Okifo, O., Robbins, K.J., Motwani, T., Alexandrescu, A.T. & Teschke, C.M. & (2016) “Contextual role of a salt-bridge in the phage P22 coat protein I-domain.”, J. Biol. Chem. 291, 11359-11372.
Matousek, W.M., Ciani, B., Fitch, C.A., Garcia-Moreno E., B., Kammerer, R.A. & Alexandrescu, A.T. (2007) "Electrostatic contributions to the stability of the GCN4 leucine zipper structure". J. Mol. Biol. 374, 206-219.
Structure of the agrin C-terminal domain
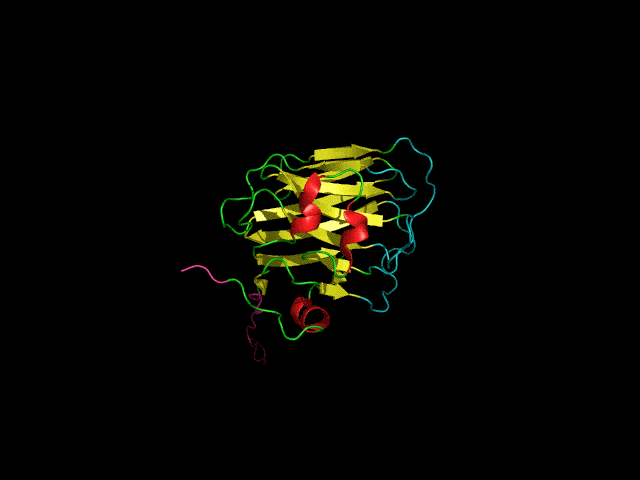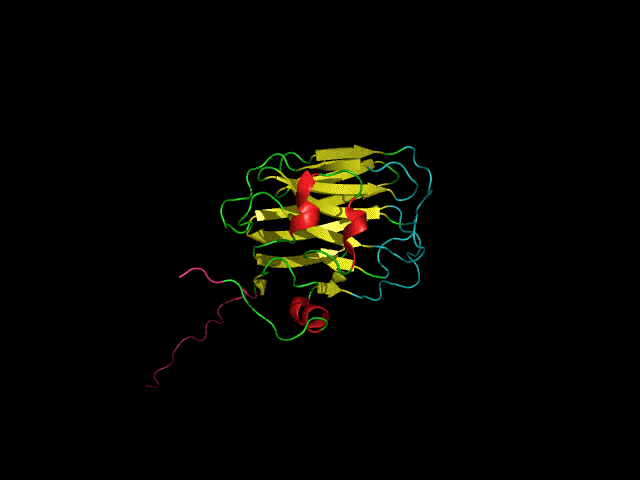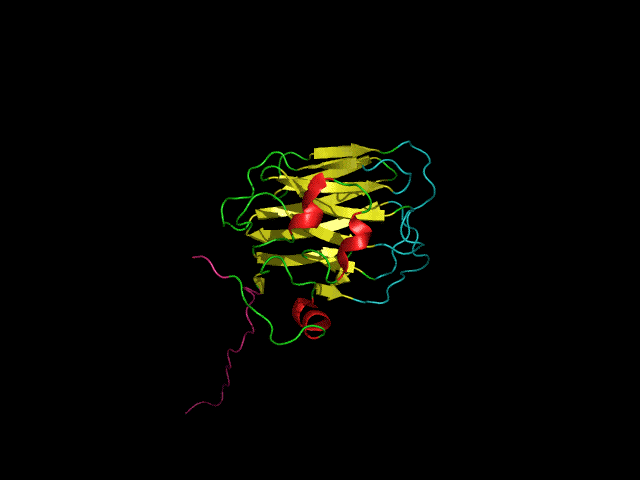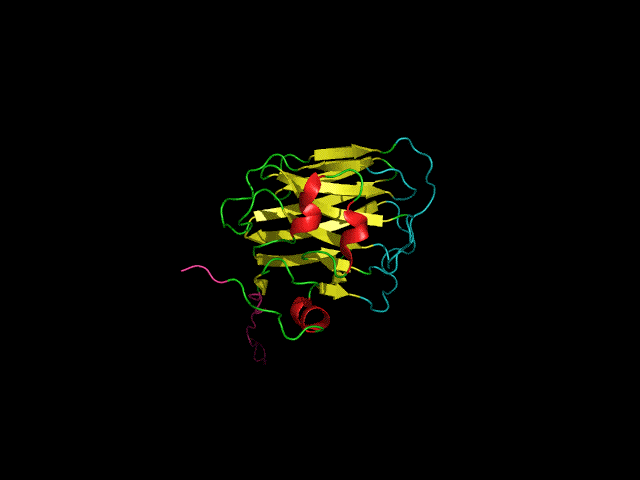
The C-terminal agrin-G3 domain (21 kDa) is involved in clustering acetylcholine receptors, a key step in the differentiation of synapses between nerves and muscles. The structure of the domain consists of 13 beta strands arranged in two sheets that form a beta-jellyroll. Our work on the structure and dynamics of this domain showed that while the beta-sheet structure (yellow) is rigid the loops responsible for the activity of the domain (light blue) are flexible. We showed that the domain binds calcium. Calcium, which is required for clustering activity reduces but does not eliminate flexibility in the 'active site' loops. More recently we showed that the domain binds carbohydrates such as sialic acid and glycosaminoglycans (heparin & heparan sulfate) which are likely to have important roles in agrin's function
Stetefeld, J., Alexandrescu A. T., Maciejewski, M. W., Jenny, M., Rathgeb-Szabo, K., Schulthess, T., Landwehr, R., Frank, S., Ruegg, M.A., & Kammerer, R.A. (2004) "Modulation of agrin function by alternative splicing and Ca2+ binding". Structure 12, 503-515.
Sallum, C.O., Kammerer, R.A., & Alexandrescu, A.T. (2007) “Thermodynamic and structural studies of carbohydrate binding by the agrin-G3 domain”, Biochemistry 46, 9541-9550.
Conserved folding of proteins that share a similar structure motif
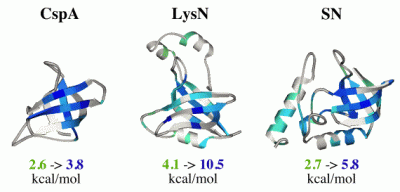 The OB-fold (oligonucleotide/oligosaccharide-binding fold) occurs in more than 80 unrelated proteins. It consists of a 5-stranded beta barrel formed from two- and three-stranded beta-sheets. We have studied the folding of three OB-fold proteins that share no detectable sequence homology: SN (staphylococcal nuclease), LysN (anticodon binding domain of Lys-tRNA synthetase) and CspA (Cold shock protein A). Mutagenesis and hydrogen exchange experiments show that the structural motif conserved between the three proteins is more resistant to unfolding than structural elements that are not conserved between the proteins. This is shown in the figure on the right where the site-specific stabilities to unfolding events that expose amide hydrogens to exchange are mapped on the structures (blue is more stable, green is less stable, white is undetermined). In fact, misfolding of the three proteins also appears to be conserved. The first three strands of beta-sheet show the highest propensity for structure under denaturing conditions, and the acid denatured forms of all three proteins aggregate through the mispairing of this partially formed unfulfilled structure. We are currently exploring the structural determinants of conserved motifs in the OB-fold.
The OB-fold (oligonucleotide/oligosaccharide-binding fold) occurs in more than 80 unrelated proteins. It consists of a 5-stranded beta barrel formed from two- and three-stranded beta-sheets. We have studied the folding of three OB-fold proteins that share no detectable sequence homology: SN (staphylococcal nuclease), LysN (anticodon binding domain of Lys-tRNA synthetase) and CspA (Cold shock protein A). Mutagenesis and hydrogen exchange experiments show that the structural motif conserved between the three proteins is more resistant to unfolding than structural elements that are not conserved between the proteins. This is shown in the figure on the right where the site-specific stabilities to unfolding events that expose amide hydrogens to exchange are mapped on the structures (blue is more stable, green is less stable, white is undetermined). In fact, misfolding of the three proteins also appears to be conserved. The first three strands of beta-sheet show the highest propensity for structure under denaturing conditions, and the acid denatured forms of all three proteins aggregate through the mispairing of this partially formed unfulfilled structure. We are currently exploring the structural determinants of conserved motifs in the OB-fold.
Alexandrescu, A. T., Gittis, A., Abeygunawardana, C., & Shortle, D. (1995) “NMR structure of a stable "OB-fold" sub-domain isolated from staphylococcal nuclease”. J. Mol. Biol. 250, 134-143.
Alexandrescu, A.T., Jaravine, V.A., Dames, S.A. & Lamour, F.P. (1999) "NMR hydrogen exchange of the OB-fold protein LysN as a function of denaturant: The most conserved elements of structure are the most stable to unfolding". J. Mol. Biol. 289, 1041-1054.
Jaravine, V.A., Rathgeb-Szabo, K., & Alexandrescu, A.T. (2000) "Microscopic stability of cold shock protein A examined by NMR native state hydrogen exchange as a function of urea and trimethylamine N-oxide". Protein Science 9, 290-301.
Alexandrescu, A.T., & Rathgeb-Szabo, K. (1999) "An NMR investigation of solution aggregation reactions preceding the misassembly of acid denatured cold shock protein A into fibrils". J. Mol. Biol. 291, 1191-1206.
Alexandrescu, A.T., Jaravine, V.A., & Lamour, F.P. (2000) "NMR evidence for progressive stabilization of native-like structure upon aggregation of acid denatured LysN". J. Mol. Biol. 295, 239-255.
Watson, E., Matousek, W.M., Irimies, E.L. & Alexandrescu, A.T. (2007) “Partially folded states of staphylococcal nuclease highlight the conserved structural hierarchy of OB-fold proteins”, Biochemistry 46, 9484-9494.
Formation of individual hydrogen bonds during folding of an alpha-helix
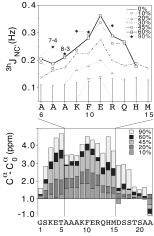
A technique developed by Stefan Grzesiek to detect scalar coupling mediated through hydrogen bonds was used to follow alpha-helix formation in the S-peptide of ribonuclease A. The results show that hydrogen bonds are not uniformly populated as alpha-helical structure forms. The hydrogen-bonds follow a stability gradient that decreases from the center to the ends of the helix. The results are roughly consistent with predictions from the Lifson-Roig theory of coil-helix transitions.
Jaravine, V.A., Alexandrescu A. T., & Grzesiek, S. (2001) "Observation of the closing of individual hydrogen bonds during TFE-induced helix formation in a peptide". Protein Science 10, 943-950.
NMR structure of a homotrimeric coiled coil
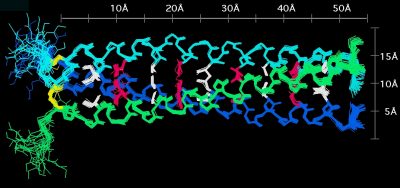 We determined the first NMR structure of a homotrimer. Homooligomers pose a particular problem for NMR because the monomer chains are magnetically equivalent. Additional approaches are thus needed to distinguish NOEs within a chain from those between chains. To solve the NMR structure of the coiled coil trimer from matrilin-1 we first obtained a highly defined structure for the monomers. We then used a self-consistent strategy to assign NOEs between chains, which were identified from isotope-filtered NOE experiments.
We determined the first NMR structure of a homotrimer. Homooligomers pose a particular problem for NMR because the monomer chains are magnetically equivalent. Additional approaches are thus needed to distinguish NOEs within a chain from those between chains. To solve the NMR structure of the coiled coil trimer from matrilin-1 we first obtained a highly defined structure for the monomers. We then used a self-consistent strategy to assign NOEs between chains, which were identified from isotope-filtered NOE experiments.
Dames, S.A., Wiltscheck, R., Kammerer, R.A., Engel, J., & Alexandrescu, A.T. (1998) "NMR structure of a parallel homotrimeric coiled coil". Nature Struct. Biol
Residual dipolar couplings in denatured proteins
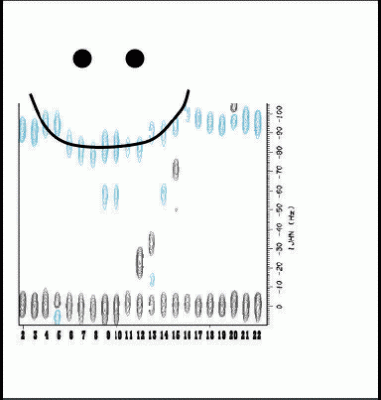
We showed how dipolar couplings can arise from localized anisotropic structure in denatured proteins, that dipolar couplings are biased towards the most anisotropic structures in the denatured state ensemble, and examined how dynamics and changes in the distributions of conformers in the denatured state ensemble affect couplings.
Alexandrescu, A.T., & Kammerer, R.A. (2003) “Structure and disorder in the ribonuclease S-peptide probed by NMR residual dipolar couplings”. Protein Sci. 12, 2132-2140.
Sallum, C.O., Martel, D.M., Fournier, R.S., Matousek, W.M & Alexandrescu, A.T. (2005) “Sensitivity of NMR residual dipolar couplings to perturbations in folded and unfolded staphylococcal nuclease”. Biochemistry 44, 6392-6403.
.
.
Dynamics in denatured and partially folded states
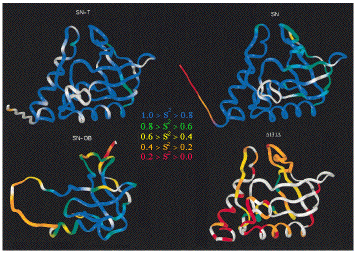 15N relaxation measurements have been used to investigate dynamics in a number of equilibrium folding intermediates. The figure shows four forms of the enzyme staphylococcal nuclease with different levels of cooperatively stabilized structure. SN-T is the nuclease in a ternary complex with Ca2+ and the inhibitor pdTp, which stabilize the protein. SN is the wild type protein. SN-OB is a folded fragment of the nuclease that is missing 1/3 of the chain from the C-terminus (see below). D131D is a highly denatured fragment of nuclease with near-random-coil NMR spectra. Blue colors indicate low order parameters (related to the amplitude of motion) and rigid structure, red colors indicate large order parameters and flexibility. As the stability of the native state stability decreases, the main-chain is subject to increasingly larger amplitude motions and the dynamic properties of the chain become more heterogeneous. These results paint a picture of unfolding as a fragmentation (or shattering) of the native state structure, and of folding as an accretion of structure in which the mobility of the main chain is frozen-out and initially separate structural elements become increasingly interdependent in order to achieve the maximum cooperativity and stability of the native state.
15N relaxation measurements have been used to investigate dynamics in a number of equilibrium folding intermediates. The figure shows four forms of the enzyme staphylococcal nuclease with different levels of cooperatively stabilized structure. SN-T is the nuclease in a ternary complex with Ca2+ and the inhibitor pdTp, which stabilize the protein. SN is the wild type protein. SN-OB is a folded fragment of the nuclease that is missing 1/3 of the chain from the C-terminus (see below). D131D is a highly denatured fragment of nuclease with near-random-coil NMR spectra. Blue colors indicate low order parameters (related to the amplitude of motion) and rigid structure, red colors indicate large order parameters and flexibility. As the stability of the native state stability decreases, the main-chain is subject to increasingly larger amplitude motions and the dynamic properties of the chain become more heterogeneous. These results paint a picture of unfolding as a fragmentation (or shattering) of the native state structure, and of folding as an accretion of structure in which the mobility of the main chain is frozen-out and initially separate structural elements become increasingly interdependent in order to achieve the maximum cooperativity and stability of the native state.
Alexandrescu, A.T., Jahnke, W., Wiltscheck, R., & Blommers, M.J.J (1996) "Accretion of structure in staphylococcal nuclease: An 15N NMR relaxation study". J. Mol. Biol. 260, 570-587.
NMR structure of a sub-domain from a cooperatively folded protein
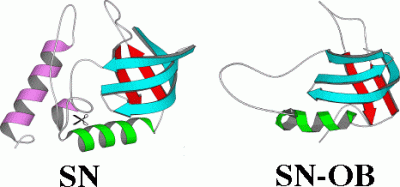
Staphylococcal nuclease folds with the typical all-or none cooperativity of a single domain protein. In the presence of the two rare global suppressor mutations V66L and G88V (analogous mutations occur for thermostable nucleases found in nature) a 1-103 fragment of nuclease remains folded in the absence of the last 1/3 (55 residues) of the sequence (purple segment). The portion of the structure that remains folded corresponds to the conserved OB-fold motif found in a variety of protein structures. The C-terminal 1/3 of the sequence is certainly not dispensable since it contains part of the enzyme's active site. The fragment has a 1000-fold lower nuclease activity than the wild type, but still classifies as an enzyme (reaction rate is 10^12 fold higher with the fragment). The fact that part of the polypeptide chain of this single-domain protein can be deleted challenges two-state models of protein folding. It also suggests mechanisms by which protein structures can evolve by assimilating pre-existing structural motifs; sacrificing the independence of sub-domains in order to achieve the maximum cooperativity and stability of the new integrated structure.
Alexandrescu, A. T., Gittis, A., Abeygunawardana, C., & Shortle, D. (1995) “NMR structure of a stable "OB-fold" sub-domain isolated from staphylococcal nuclease”. J. Mol. Biol. 250, 134-143.
A structural transition involved in bacterial signal transduction